

Introduction: While advances in front-line conventional chemotherapy have increased the likelihood of attaining remission in pediatric AML, relapse rates remain high (25-35%), and novel therapies are needed (Zhang, Savage et al. 2019). The clinical and molecular heterogeneity of AML makes it complex to study and creates challenges for the development of novel therapies (Bolouri, Farrar et al. 2018). It is important to identify cells and pathways underlying relapse to facilitate development of novel therapies. Single-cell RNA Sequencing (scRNA-Seq) allows in-depth analysis of the heterogeneous AML landscape to provide a detailed view of the tumor microenvironment, revealing populations of blasts and immune cells which may be relevant to relapse or complete remission.
Methods: We analyzed ~36,500 cells from 14 pediatric AML bone marrow samples in our institutional biorepository, spanning different AML subtypes and 3 healthy children to generate a comprehensive scRNA-Seq landscape of immature AML-associated blasts and microenvironment cells. Samples collected at the time of diagnosis (Dx), end of induction (EOI), and relapse (Rel) were used to generate scRNA-Seq data using a droplet-based barcoding technique (Panigrahy, Gartung et al. 2019). After normalization of scRNA-Seq data, the cell clusters were identified using principal component analysis and Uniform Manifold Approximation and Projection (UMAP) approach (Becht et al, 2018). Differential expression, pathways and systems biology analysis between relapsed and remission patients reveal differences for specific cell clusters (Panigrahy, Gartung et al. 2019). To determine the clinical outcome association of our AML blast specific markers, survival analysis was performed on AML TARGET data (https://ocg.cancer.gov/programs/target) using cox proportional hazard survival approach. To characterize AML blast cells with high accuracy, we used support vector machine (SVM), an Artificial Intelligence based feature extraction and model development approach (Bhasin, Ndebele et al. 2016).
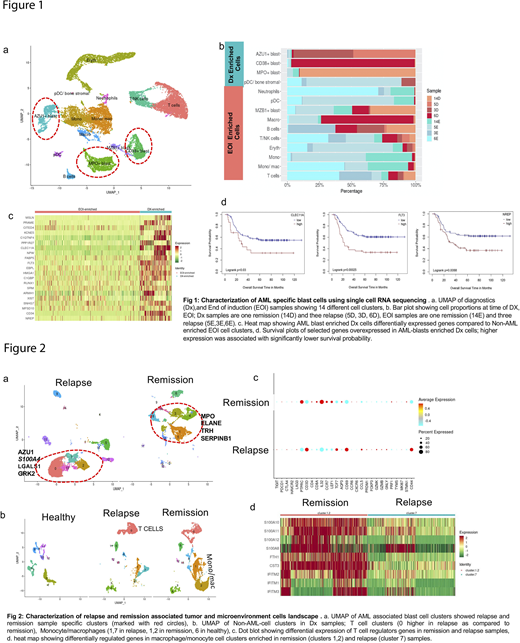
Results:ScRNA-Seq analysis of paired Dx and EOI samples using UMAP identified three blast cell clusters with significant gene expression differences among different patients, indicating heterogeneity of AML blast cells (Fig 1a, b). Comparative analysis of the three Dx enriched blast cell clusters with other cells identified a "core blast cell signature" with overexpression of genes like AZU1, CLEC11A, FLT3, and NREP (Fig 1c). These core AML-blast genes were linked to significant activation of the Wnt/Ca2+, Phospholipase C, and integrin signaling pathways (Z score >2 and P-value <.001). A subset of AML blast-specific genes also depicted significant association with poorer overall survival (CLEC11A Hazard ratio (HR)=1.9 ;95% CI=1.1-3.4;log-rank P=.03, FLT3 HR=2.4(1.5-3.9);P<0.001,NREP HR=1.9(1.2-3.1); P<0.008) (Fig 1d). Furthermore, we developed a highly sensitive 7-gene AML blast cell signature that distinguished AML blasts from normal myeloblasts and other hematopoietic cells (Area Under Curve of 0.94) using SVM.
The scRNA-Seq of AML specific blast cells from relapsed and remission samples exhibited a different clustering pattern indicating different transcriptome landscapes. Relapse-associated AML cell clusters expressed high levels of AZU1, S100A4, LGALS1, and GRK2 genes (Fig 2a). Analysis of the non-AML tumor microenvironment demonstrated enrichment of T/NK in relapsed samples, with differential expression of T cell regulatory/activation genes (Fig 2b, c). ScRNA-Seq showed enrichment of monocyte/macrophage cell clusters in remission samples with distinct relapse- and remission-specific clusters. Remission associated macrophage/monocyte clusters showed overexpression of S100A10, FTH1, CST3 and IFITM2 genes (Fig 2d). Similarly, enrichment of T cell and monocyte/macrophage clusters was observed in relapse and remission samples respectively during EOI.
Conclusions: Using single cell transcriptomics we developed a novel potential gene signature to characterize heterogenous AML blast populations with high sensitivity. These genes and the pathways they regulate implicate potential therapeutic targets in pediatric AML. Single cell transcriptome analysis also enabled identification of cell clusters with modulated gene expression at both Dx and EOI that may be useful in predicting relapse/remission.
Bhasin:Canomiiks Inc: Current equity holder in private company, Other: Co-Founder.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal